Pediatr Integral 2018; XXII (6): 282 – 293
Meningoencefalitis viral
Introducción
Las infecciones virales del sistema nervioso central (SNC) pueden manifestarse como un cuadro meníngeo, produciendo la sintomatología clásica de: fiebre, cefalea, náuseas, vómitos, fotofobia y rigidez nucal. También pueden manifestarse en forma de: inflamación del parénquima, encefalitis, presentando en mayor o menor grado disfunción neurológica, disminución del nivel de conciencia, letargia, alteraciones en la personalidad o el comportamiento, convulsiones o focalidad neurológica. Hablamos de meningoencefalitis cuando existe una inflamación, tanto de las meninges como del parénquima cerebral, con la consiguiente suma de las potenciales manifestaciones clínicas comentadas(1-4).
Los virus son la causa más frecuente de infecciones del SNC, con muchos posibles agentes etiológicos implicados. Pueden causar enfermedad por infección directa, y replicación en SNC (encefalitis viral aguda), o por procesos mediados por mecanismos inmunológicos. Estas entidades, difíciles de diferenciar clínicamente, a menudo presentan diferencias radiológicas muy útiles en su diagnóstico diferencial(1-3,5).
Epidemiología
Los virus son responsables de la inmensa mayoría de infecciones del SNC. Estas infecciones presentan dos picos de máxima incidencia en Pediatría: menores de un año y niños entre 5-10 años. Es difícil conocer la epidemiología exacta, por la gran variedad de síntomas que pueden producir, lo que hace que la presentación clínica, en ocasiones, no sea específica y pueda confundirse con otras entidades. Se estima una incidencia de encefalitis de 1,5 a 7 casos /100.000 habitantes/año, sin tener en cuenta las epidemias. Las infecciones virales del SNC son más frecuentes en los meses de primavera, verano y otoño (mayor tasa de infecciones enterovirales y de virus transmitidos por artrópodos), y parecen ser ligeramente más frecuente en niños que en niñas (en torno al 55%)(1-3,5-7).
Etiología
La familia de enterovirus (EV), que incluye: poliovirus, echovirus, coxsackievirus, rhinovirus y los propios enterovirus, producen en la mayoría de las ocasiones infecciones asintomáticas. Sin embargo, son la causa más común, tanto de meningitis como de encefalitis viral en la edad pediátrica (85-95% en meningitis y 10-20% encefalitis virales, respectivamente). Aunque algunos serotipos mantienen su prevalencia durante todo el año, generalmente producen brotes durante los meses de verano y otoño. Los poliovirus asocian en un bajo porcentaje de sus infecciones (?1%) parálisis flácida y poliomielitis paralítica; aunque actualmente, el virus solo circula en unos pocos países de África y Asia (Nigeria, Pakistán y Afganistán). El enterovirus A-71 (EV71) es considerado como el EV no poliovirus más neuropatogénico, puede afectar al SNC produciendo: meningitis, encefalitis, romboencefalitis y, en raras ocasiones, parálisis flácida o edema pulmonar neurogénico(3,5,13).
En la primavera de 2016, se registró un brote epidémico de infección por EV71 en Cataluña, con clínica neurológica asociada (romboencefalitis y, ocasionalmente, mielitis), siendo el mayor registrado en los últimos años en Europa, y no coincidiendo con brotes similares en países próximos. En este brote, que tuvo su pico a mediados del mes de mayo, se registraron casos procedentes de las cuatro provincias catalanas, y se vieron afectados un total 105 niños(13).
Los paraechovirus (PeV), especie muy similar a los EV, producen habitualmente cuadros respiratorios o gastrointestinales autolimitados, aunque pueden producir: meningitis, encefalitis y cuadros de sepsis en menores de 3 meses, sobre todo, el PeV3. También son más frecuentes en verano y otoño(8,10,12).
Los virus de la familia herpes son frecuentemente responsables de infecciones del SNC, como el virus herpes simple (VHS), que produce infecciones graves, siendo la causa más común de encefalitis necrotizante (>85%). Cuando la infección por VHS se adquiere a través del canal del parto, puede causar cuadros de meningoencefalitis en neonatos. El virus varicela zóster (VVZ) puede producir: encefalitis, meningitis, cerebelitis (afectación más común) o mielitis. Suele asociar vasculitis y lesiones hipóxico-isquémicas. Otros virus de esta familia como: citomegalovirus (CMV), virus de Ebstein-Barr (VEB) y virus herpes humanos 6 y 7 (VHH-6 y VHH-7) también pueden infectar el SNC, aunque mucho menos frecuentemente en pacientes inmunocompetentes(1,3,5,7,9,14).
Los arbovirus son virus transmitidos por la picadura de artrópodos, que también pueden afectar al SNC, aunque en la mayoría de las ocasiones producen infecciones asintomáticas. Presentan distribución mundial, con diferencias según áreas geográficas, y mayor incidencia en climas tropicales. En climas templados son más frecuentes durante el verano-otoño. En Europa, los arbovirus más frecuentes son: el virus de la encefalitis por picadura de garrapata (tick borne virus) o encefalitis centroeuropea, que afecta a más de 20 de los 30 países de la Unión Europea y Rusia (Fig. 1), y el West Nile Virus, endémico en el este y norte de África, Asia, Australia y norte y sur de América, del que se han documentado casos en el este y sur de Europa.

Figura 1. Casos de encefalitis centroeuropea declarados por 100.000 habitantes, en 2015, ECDC(20).
Otros arbovirus infrecuentes en nuestro medio, que deben ser considerados en viajeros e inmigrantes de zonas endémicas son: el virus de la encefalitis japonesa (endémica en zonas rurales del sudeste asiático, China, Nepal e India), el virus de la encefalitis de Sant Louis, el virus de La Crosse y el virus de la encefalitis equina en América o los virus dengue, chikungunya y zika, localizados en áreas tropicales, aunque estos últimos no suelen producir encefalitis(1,3,5,15).
La rabia, aunque actualmente erradicada de animales terrestres en la península ibérica, también puede producir infecciones letales del SNC; en nuestro entorno, tienen riesgo de sufrirla aquellos niños que viajan a áreas hiperendémicas o son mordidos por murciélagos (situación poco frecuente en nuestro medio)(3).
Algunos virus respiratorios como el de la gripe (mayor riesgo en infección por gripe A), metapneumovirus, virus respiratorio sincitial (VRS) o adenovirus, además de virus para los que habitualmente existe la vacunación (sarampión, parotiditis, rubéola), aún prevalentes en países donde no hay vacunación universal, o en pacientes no vacunados, y el virus de la inmunodeficiencia humana (VIH), también pueden ser causa de infección viral del SNC(3,7).
En muchas de las encefalitis con etiología viral sospechada no se identifica la causa (25-50%). Estos casos podrían deberse a mecanismos patogénicos inmunológicos, infecciones causadas por virus emergentes (Chandipura o Nipah virus en el sudeste asiático, Powassan en Norteamérica, o Hendra en Australia) o virus transmitidos desde otras áreas geográficas, no identificados(1-6,9,16).
Fisiopatología
La mayoría de virus que infectan el SNC, inicialmente colonizan las vías respiratorias o el tracto gastrointestinal, consiguen atravesar la barrera epitelial y llegar a los ganglios linfáticos locales, donde se replican. Posteriormente, se produce una viremia con diseminación del virus a otros órganos, principalmente el sistema reticuloendotelial. Allí se replica de nuevo y da lugar a una segunda viremia, generalmente mayor que la inicial, que suele asociar la aparición de signos y síntomas de la infección. El paso de virus al SNC puede darse durante la primera o la segunda viremia. Se desconocen los mecanismos específicos por los que el virus es capaz de alcanzar el SNC por vía hematógena(1,6). En el caso del VHS, se especula la posibilidad de diseminación a través de la placa cribiforme, desde la infección de la mucosa nasofaríngea o por diseminación neurógena(1,8,10).
Los EV y virus de la familia herpes se transmiten entre humanos (único reservorio conocido) por contacto directo (VHS, EV) o vía fecal-oral (EV, PeV). Algunos serotipos de EV pueden transmitirse también por inhalación. En el caso de los arbovirus, la transmisión se produce por la picadura de artrópodos (mosquitos o garrapatas)(1,8,15).
El periodo de incubación es variable, de 3-6 días en EV, 2-12 en VHS o 4-28 en la encefalitis centroeuropea, pudiendo ser de hasta meses en el caso del virus de la rabia(8,15).
Al parecer, los linfocitos T CD8+ desempeñan un papel importante en el desarrollo de la inmunidad celular, y en mecanismos inmunopatológicos que pueden causar daño cerebral. Algunas mutaciones en los receptores de membrana del sistema inmune, como el TLR-3, también parece ser un factor de riesgo para el desarrollo de encefalitis viral (principalmente causada por VHS-1) en humanos(1,8,11). Por ello, la frecuencia de meningoencefalitis virales es mayor en el paciente inmunodeprimido(1).
Clínica
Las manifestaciones clínicas de las meningitis virales son similares a las meningitis ocasionadas por infección bacteriana, con síntomas como: fiebre, cefalea, vómitos, rigidez nucal y fotofobia, aunque normalmente menos graves. A menudo, la cefalea y la fotofobia son los síntomas más evidentes, especialmente en las causadas por EV(3,7,15).
En la encefalitis existe disfunción neurológica, que puede presentarse con muy diversos signos y síntomas como: disminución del nivel de conciencia, letargia, alteraciones de la personalidad o el comportamiento, convulsiones, focalidad neurológica, temblor, ataxia, disfagia o disartria, en función del agente viral responsable y la región afectada (Tabla I).
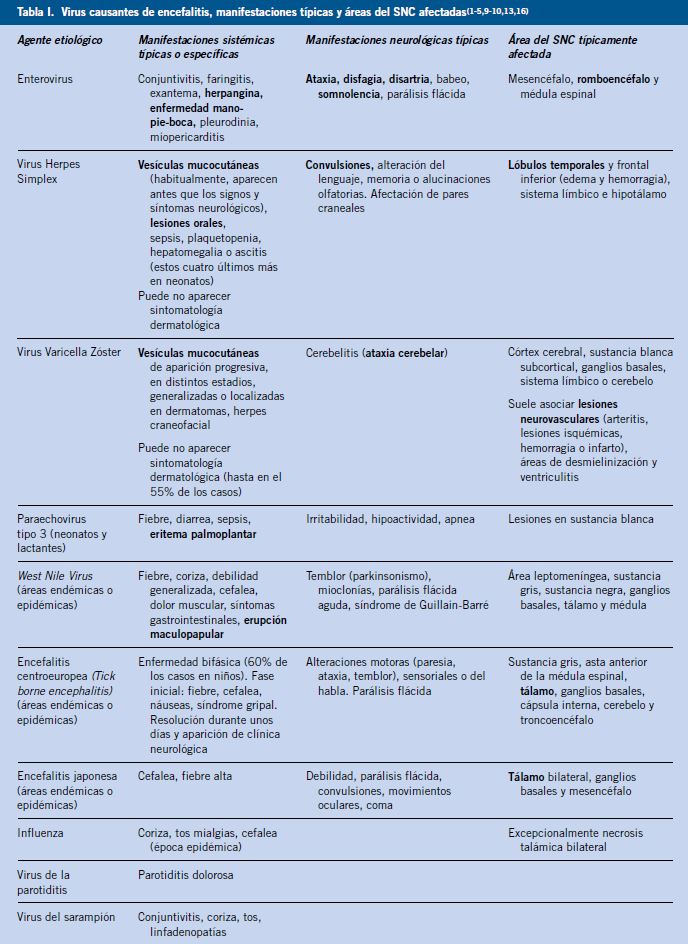

En niños mayores de 5 años y adolescentes, puede ocasionar: síntomas psiquiátricos, labilidad emocional, alteraciones en los movimientos, ataxia o estupor. En casos graves, puede producir: estatus epiléptico, edema cerebral, síndrome de secreción inadecuada de hormona antidiurética y fallo cardiorrespiratorio. La inflamación del SNC puede darse a nivel cerebral; del tronco del encéfalo (rombencefalitis o encefalitis del troncoencéfalo), pudiendo producir mioclonía, temblor, ataxia, alteración de los pares craneales o alteraciones respiratorias; a nivel medular (mielitis), con debilidad, disfunción vesical, parálisis flácida y alteración de los reflejos; o a nivel periférico, en las raíces nerviosas (radiculitis) produciendo debilidad, disestesias y alteración de los reflejos(3,7,9).
Además de la clínica por afectación del SNC, suelen presentar clínica sistémica inespecífica característica de la infección viral, como: anorexia, náuseas, exantema, mialgias, sintomatología respiratoria. En algunos casos, existe clínica más específica del agente viral responsable como se recoge en la tabla I(2,3,7,9).
El desarrollo de formas más graves o crónicas de enfermedad depende también de factores del huésped, siendo la edad y la presencia de trastornos del sistema inmunitario posiblemente los más importantes(1,3).
Los neonatos pueden presentar sintomatología mínima o inespecífica, como: fiebre (variable), convulsiones, rechazo de la alimentación, vómitos, diarrea, erupción cutánea, síntomas respiratorios, irritabilidad, letargia y abombamiento de la fontanela. Este grupo presenta mayor facilidad para desarrollar infección diseminada, pudiendo desarrollar: neumonía, hepatitis, miocarditis, enterocolitis necrotizante, coagulación intravascular diseminada u otros hallazgos de sepsis. Tienen especial riesgo de padecer formas graves de la enfermedad (del SNC y sistémicas), sobre todo en infecciones causadas por VHS o EV(1-3,17).
Respecto a los pacientes con un trastorno inmunitario, son más propensos a desarrollar encefalitis por CMV, VHH6, VVZ o EV. Es especialmente interesante conocer la relación de la meningoencefalitis crónica por EV y la presencia de defectos de producción de anticuerpos, especialmente aquellos con agamma-globulinemia ligada al cromosoma X. Su pronóstico es desfavorable, con evolución a un cuadro de demencia progresivo sin tratamiento, que a menudo lleva al fallecimiento del paciente por complicaciones asociadas(3,5,7-9).
Evaluación y pruebas complementarias
Las infecciones del SNC precisan de una rápida evaluación y tratamiento. Es importante definir el cuadro clínico (meningitis, meningoencefalitis, encefalitis…) e intentar identificar la etiología lo antes posible, ya que puede ser importante para el manejo del paciente, la posible profilaxis necesaria y otras intervenciones de salud pública. El inicio precoz del tratamiento mejorará notablemente el pronóstico y la obtención de los estudios analíticos ha de realizarse tan pronto como sea posible(2,3,7).
Anamnesis
Se debe preguntar por: viajes, infecciones previas, medicación, exposiciones a enfermos, animales, insectos, comida contaminada y revisar el calendario vacunal, así como por otros antecedentes personales y familiares. En lactantes y neonatos, se ha de preguntar por la historia perinatal. Es muy importante conocer la epidemiología de la región(1,2,5,15).
Examen físico
Deberemos explorar signos meníngeos y realizar una evaluación neurológica: focalidad, orientación, motor, sensitivo, pares craneales, cerebelo y reflejos. La escala de Glasgow puede ser útil para cuantificar y evaluar la evolución del nivel de conciencia. La exploración física puede ayudar a identificar el agente etiológico (Tabla I)(2,7-9).
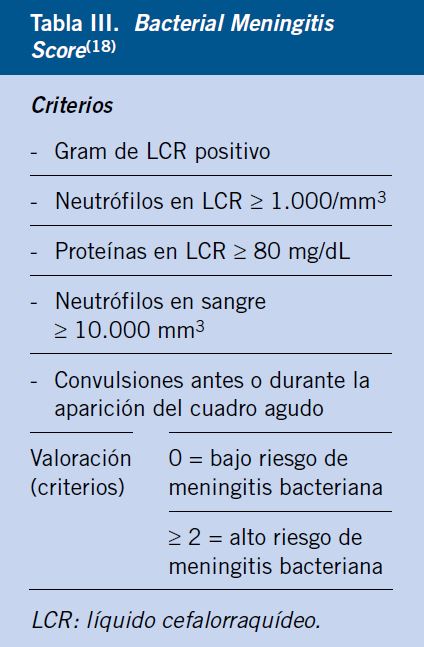
Se debe realizar un hemograma completo, bioquímica con iones, glucosa, función renal y hepática, además de recogida de hemocultivo si no puede descartarse etiología bacteriana. Generalmente, los resultados de estos estudios son inespecíficos en esta clase de infecciones, aunque algunos hallazgos pueden orientar a la etiología, como la aparición de: neutropenia, trombocitopenia, elevación de transaminasas o hiponatremia en la infección neonatal por VHS. Los valores de mediadores inflamatorios, como la proteína C reactiva o la procalcitonina, así como el recuento leucocitario pueden ayudar a distinguir meningitis bacteriana de vírica, con distintos scores clínico-analíticos, como el de Boyer o el Bacterial Meningitis Score (Tablas II y III)(2,7,18-19).

Estudio de líquido cefalorraquídeo (LCR)
Se recogerá muestra en todos los pacientes con sospecha de afectación infecciosa del SNC, salvo en caso de sospecha de lesión ocupante de espacio –en los que previamente se realizará prueba de imagen–, plaquetopenia o coagulopatía graves(1-2). Las infecciones virales del SNC suelen tener presión de apertura normal o moderadamente elevada. Se debe realizar: recuento celular, estudio de las características bioquímicas (glucosa y proteínas), tinción de Gram, cultivo bacteriano y estudio molecular –reacción en cadena de polimerasa (PCR)– para VHS y EV, así como otras pruebas específicas en función de la sospecha etiológica. Además, a menudo es útil reservar una muestra adicional de LCR congelada por si en el futuro se necesitan realizar más estudios. La negatividad de los estudios realizados en LCR, no siempre descarta la enfermedad. Así, por ejemplo, en pacientes con alta sospecha de infección por VHS y PCR inicial negativa, se recomienda repetir punción lumbar en los siguientes 1-3 días tras el inicio de tratamiento, ya que la PCR puede ser negativa en las primeras horas o días de la enfermedad, positivarse posteriormente y permanecer así hasta pasada la primera semana de tratamiento. El aislamiento de IgM específica en LCR es diagnóstico de enfermedad (no atraviesa barrera hematoencefálica) y puede usarse para el diagnóstico de algunas entidades, como las encefalitis por arbovirus, para las que las pruebas de PCR no están tan desarrolladas o tienen peores resultados. Las características del LCR en encefalitis, meningoencefalitis o meningitis virales pueden ser similares, pudiendo además solaparse con las de la meningitis bacteriana (Tablas II-IV). Además, los resultados de la citoquímica del LCR no son específicos de un virus, y los pacientes con infección viral del LCR pueden tener características totalmente normales del LCR. Actualmente, hay en desarrollo paneles de PCR múltiple para muestras de LCR, que permiten realizar el diagnóstico de infecciones virales, bacterianas y fúngicas del SNC en paralelo. Según el grado de sospecha, los estudios de anticuerpos (encefalitis autoinmune) se solicitarán a la vez que los microbiológicos(1-3,7-9,15-16,18-19).

Identificación del patógeno específico en otros tejidos o secreciones
Es posible aislar el patógeno en otro lugar anatómico distinto del SNC. Puede ser en aspirado nasofaríngeo o heces, mediante cultivo o PCR, como en el caso de romboencefalitis o mielitis por EV, ya que algunas cepas como EV71, se aíslan difícilmente en LCR. En caso de lesiones cutáneas compatibles con infección por VHS, VVZ u otros virus, se debe realizar estudio molecular mediante PCR o inmunofluorescencia directa de las lesiones. En menores de 6 semanas con sospecha de enfermedad diseminada por VHS, se debe realizar PCR de VHS en sangre. En pacientes inmunocomprometidos (incluidos menores de 3 meses), es importante la recogida de muestras de sangre y orina para realizar estudios virales, así como la búsqueda de patógenos no virales. También en caso de sospecha de arbovirus, puede buscarse en sangre, aunque no son fáciles de aislar por la breve duración de la viremia. Los resultados de estas pruebas deben interpretarse en función de la clínica y epidemiología; de tal forma que, en ocasiones, resultados positivos pueden no tener significación, o no presentar relación con la afectación del SNC. Cuando se aísla enterovirus en paciente con encefalitis (en muestra de LCR o en otros tejidos o secreciones), está indicado remitir la muestra al Laboratorio Nacional de Microbiología (Majadahonda, Madrid) para el genotipado de la cepa. La biopsia cerebral está indicada en raras ocasiones, limitándose su realización a situaciones muy específicas en las que el paciente continúa empeorando a pesar del tratamiento(2,10,13).
Serologías
Su utilidad es relativa, mayor cuanto más infrecuente sea el agente etiológico sospechado (sarampión, parotiditis, varicela, arbovirus, VIH). Al igual que la identificación de patógenos en otros tejidos o secreciones, deben valorarse en conjunto con otros resultados, las características clínicas y epidemiológicas del paciente. Se ha de tener en cuenta que muchos arbovirus presentan reactividad cruzada en estas pruebas y, en ocasiones, requieren de pruebas de confirmación adicionales. Es conveniente repetir el estudio tras 4 semanas para ver la evolución de los títulos de anticuerpos. En muchas ocasiones, la serología es más útil para un diagnóstico retrospectivo que en el momento agudo(1-3,5,15).
Neuroimagen
Su realización está indicada en caso de sospecha de encefalitis y se tiene que realizar antes de la punción lumbar en pacientes con signos o síntomas de: aumento de la presión intracraneal (alteración del estado mental, edema de papila, déficit neurológico focal), inmunodeficiencia, alteraciones previas en SNC (fístula, hidrocefalia, lesión ocupante de espacio), traumatismo del SNC, sospecha de infección parameníngea o tumor. La técnica de elección para la evaluación de la encefalitis es la resonancia magnética (RM), con mayor sensibilidad y especificidad que la tomografía computarizada (TC), pero en caso de no disponer de RM se recomienda realizar una TC con y sin contraste. Cualquier deterioro neurológico agudo requiere de la repetición de pruebas de imagen para descartar complicaciones. En recién nacidos y lactantes pequeños, se puede valorar la realización de ecografía transfontanelar como prueba inicial. La encefalitis viral puede producir edema cerebral y afectación de las distintas áreas encefálicas en función del virus responsable (Tabla I)(1-3,15,17).
Electroencefalograma (EEG)
Está indicada su realización en encefalitis y se debe realizar lo antes posible. Se encuentra alterado en alrededor del 90% de los casos, la mayoría con hallazgos inespecíficos como enlentecimiento general de la actividad. Tiene especial indicación en pacientes sin actividad convulsiva, que estén confusos, obnubilados o comatosos, para detectar crisis no convulsivas no evidentes clínicamente. La afectación temporal es sugestiva de infección por VHS. En caso de encefalitis con afectación troncoencefálica, puede resultar normal o moderadamente alterado. La mayor alteración del EEG no se correlaciona con la gravedad de la enfermedad, sin embargo, una rápida normalización sí que se relaciona con un mejor pronóstico(1-2).
Diagnóstico
El diagnóstico diferencial entre meningitis y encefalitis se hará en función de los signos y síntomas de disfunción neurológica e inflamación del parénquima cerebral. Sospecharemos la etiología viral por los datos epidemiológicos, clínicos y los estudios iniciales del LCR, ya que a menudo las características clínicas no permiten distinguir entre infección viral o bacteriana. Las etiologías no virales de meningoencefalitis que requieren tratamiento específico se han de considerar en niños con estudios de LCR negativos(1-3,9).
Se puede realizar el diagnóstico provisional de meningitis viral en función de los hallazgos clínicos y características del LCR (Tabla IV), datos epidemiológicos o mejoría de la sintomatología tras la realización de la punción lumbar. El diagnóstico de confirmación de meningitis viral se realiza con cultivos bacterianos negativos y la identificación del virus mediante PCR en LCR (o biopsia cerebral en casos excepcionales). El diagnóstico de meningitis viral permite informar más propiamente con respecto al posible pronóstico, disminuir el uso de antibióticos, disminuir el tiempo de ingreso hospitalario y tomar medidas epidemiológicas dirigidas si son necesarias(1-3).
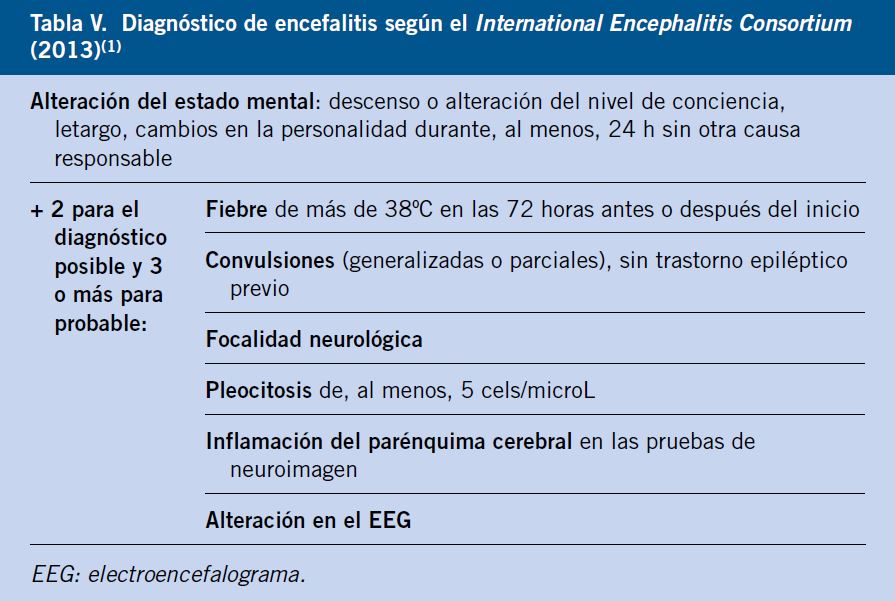
El diagnóstico de encefalitis viral requiere de los siguientes 3 criterios:
1. Disfunción neurológica evidenciada.
2. Inflamación del SNC (prueba de neuroimagen o hallazgos en EEG).
3. Exclusión de otras causas que puedan justificarlo.
La identificación del patógeno confirma el diagnóstico clínico, sin embargo, como se ha comentado, en un porcentaje importante de los casos de encefalitis no se encuentra el agente etiológico, pudiendo hacerse diagnóstico de presunción en función de los hallazgos clínicos y la exclusión de otras causas de encefalopatía.
El rápido diagnóstico por neuroimagen y técnicas de detección molecular es importante para el inicio de la terapia dirigida(1-3).
Diagnóstico diferencial
Infección del SNC por otros microorganismos
La pleocitosis en LCR con predominio monocítico no es exclusiva de la infección viral, así como los valores normales de glucorraquia o proteinorraquia, que pueden verse en infecciones bacterianas muy precoces, abortadas o en la meningoencefalitis tuberculosa(7).
Encefalitis autoinmunes
Producidas por la respuesta inmune a un desencadenante antigénico (infección viral, vacunación…). Cada vez responsables de más causas reconocidas de encefalitis en niños, pudiendo ser responsable de muchas de las encefalitis no diagnosticadas. Clínicamente, son polisintomáticas y, a veces, difíciles de diferenciar de la encefalitis viral aguda, aunque no suelen producir fiebre al debut, y se presentan de forma subaguda (Tabla I). El LCR puede ser normal o con pleocitosis de predominio linfocítico e hiperproteinorraquia. Las más frecuentes en Pediatría son las asociadas a anticuerpos anti receptor de N-metil-D aspartato (anti-NMDA), que pueden aparecer desencadenadas por encefalitis por VHS (produciendo síntomas neurológicos prolongados o atípicos tras el control inicial de la infección viral). Aunque estas encefalitis pueden recurrir, suelen hacerlo con formas monosintomáticas y de menor gravedad. En menores de 12 años, es poco frecuente la asociación de estos cuadros con neoplasias, que sí se dan en la edad adulta(1-2,4-5,9,16).
Encefalopatía por: intoxicación, enfermedad metabólica, hipoxia, isquemia, disfunción orgánica o por infección sistémica
Caracterizadas por alteración de la función neuronal sin proceso inflamatorio local. No suelen asociar fiebre, su presentación es gradual y, en ocasiones, fluctuante, sin pleocitosis en LCR ni cambios evidentes en neuroimagen en la fase aguda. Pueden producir alteraciones en la bioquímica sanguínea (hipoglicemia, acidosis e hiperamoniemia). Los estudios metabólicos o de tóxicos específicos permitirán el diagnóstico etiológico(2,9,17).
Otros
Traumatismo craneal, hemorragia, tumor en SNC, así como algunas infecciones congénitas. En estos casos, es preferible la valoración con TC a RM(1-3).
Tratamiento
Las infecciones virales del SNC, sobre todo la encefalitis, es una emergencia vital que requiere de la rápida estabilización, ingreso, tratamiento antimicrobiano empírico, si no se puede descartar la etiología bacteriana o se sospecha una infección por VHS, y medidas de soporte. Asimismo, son criterios de ingreso en las meningitis virales: la clínica de sepsis, una etiología no aclarada, requerimientos de hidratación o analgesia para control del dolor, y para algunos autores la edad menor de un año, estando indicado en estos casos, el inicio de tratamiento antimicrobiano empírico; el resto de meningitis virales pueden ser controladas de forma ambulatoria(1-3).
Tratamiento sintomático
Estabilización cardiorrespiratoria y tratamiento de las convulsiones. En caso de encefalitis grave (convulsiones, compromiso cardiorrespiratorio o neurológico grave), el paciente debe ser ingresado en una unidad de cuidados intensivos para monitorización continua. No existe consenso en cuanto a la indicación de pautar tratamiento anticomicial como profilaxis primaria o secundaria de las convulsiones, algunos autores la recomiendan en encefalitis por VHS(1-3,15).
Tratamiento empírico
Se recomienda tratamiento antibiótico empírico por vía intravenosa (cefalosporina de tercera generación y vancomicina), en espera de los resultados del estudio microbiológico en pacientes con encefalitis, y en aquellos con clínica de meningitis que además sean menores de 3 meses de edad, inmunodeprimidos, presenten un cuadro clínico grave o sospecha de infección bacteriana. El tratamiento antiviral empírico con aciclovir intravenoso se ha de iniciar precozmente en todos los casos de encefalitis; en meningitis, se recomienda en casos de LCR con características típicas de infección viral (Tabla IV), inmunosupresión o en aquellos en los que exista sospecha clínica de infección por VHS o VVZ (Tabla VI). El tratamiento empírico (antibiótico y antiviral) se interrumpirá cuando el estudio microbiológico resulte negativo (si la sospecha clínica no es alta) o se realice un diagnóstico alternativo(1-2,9,17).

La administración de corticoides inicialmente, en niños diagnosticados de meningitis aséptica no produce efectos adversos. Se ha de valorar el tratamiento empírico para otras infecciones (tuberculosis, fiebre Q, enfermedad de Lyme) en pacientes críticos con datos epidemiológicos, clínicos o analíticos sugestivos de alguna de estas enfermedades(1-3).
Tratamiento específico
En el caso de infección por VHS y VVZ, está indicado el tratamiento con aciclovir por vía intravenosa de forma precoz (Tabla VI). En la encefalitis por CMV en pacientes inmunodeprimidos, se recomienda el uso de ganciclovir. Menos clara es la indicación en encefalitis graves o progresivas por gripe con oseltamivir. En caso de infección por virus de la rabia, se realizará vacunación y administración de anticuerpos en pacientes no vacunados. Otros casos de infección viral, como EV, VEB o arbovirus no disponen actualmente de un tratamiento específico.
Tratamiento adyuvante
Los corticoides, plasmaféresis, administración de gammaglobulina inespecífica o la hipotermia terapéutica, se han utilizado en pacientes graves, aunque actualmente existe poca evidencia y se requieren más estudios para valorar su recomendación generalizada(1-3). Si los pacientes presentan encefalitis grave, alteraciones de la conciencia o sospecha de VHS o VVZ, algunos autores recomiendan asociar corticoterapia (prednisona 1 mg/kg/día o equivalente durante 3-5 días)(1,3,7,9,15).
En caso de presentar sintomatología persistente o progresiva, se deben considerar causas no virales. Realizaremos cultivos fúngicos y de micobacterias del LCR, nuevas pruebas de imagen en busca de infección parameníngea o encefalitis autoinmune, y valorar otras causas de encefalitis o encefalopatía. Puede ser útil la repetición de la punción lumbar en busca de cambios en celularidad o bioquímica o nuevos datos en el estudio microbiológico(2).
Pronóstico
Las encefalitis tienen peor pronóstico que las meningitis, que suelen tener recuperación completa, aunque durante algunas semanas tras el proceso puedan presentar síntomas como: fatiga, irritabilidad, descenso de la capacidad de concentración o incoordinación(1).
En el caso de la encefalitis, el pronóstico varía en función de la edad (peor en menores de cinco años, neonatos y lactantes), las características del paciente (inmunidad, antecedentes…), la clínica y los hallazgos al diagnóstico (peor cuanto mayor afectación al diagnóstico). La mortalidad varía en función del virus implicado (hasta del 70% en encefalitis por VHS sin tratamiento), a menudo relacionada con infección sistémica, fallo hepático o miocarditis(2-3). Los pacientes pueden presentar secuelas neurológicas como: trastornos del aprendizaje, alteraciones del comportamiento o déficits motores. Al menos, dos tercios de los pacientes con encefalitis por VHS que superan la enfermedad presentan trastornos neurológicos (incluso aquellos tratados correctamente), siendo más frecuentes la epilepsia, el retraso del desarrollo psicomotor o la parálisis(2-3).
Seguimiento
Se recomienda un seguimiento clínico estrecho en las semanas posteriores a la infección y hasta un año tras la enfermedad, incluso en los pacientes sin secuelas en la fase aguda, para la evaluación neurológica y auditiva del paciente. El seguimiento a largo plazo debería incluir evaluación del desarrollo psicomotor/neurocognitivo, sobre todo en los primeros años de escolarización, aunque no existe claro consenso ni recomendaciones en este sentido(1-2).
Prevención
Es importante la prevención de la transmisión, con medidas de higiene habituales como el lavado de manos. Se recomienda la vacunación contra algunos virus que pueden causar infección viral del SNC, como sarampión, parotiditis y polio. En caso de viaje a áreas endémicas con actividades al aire libre en áreas rurales, es recomendable la vacunación (encefalitis centroeuropea, encefalitis japonesa o rabia)(1,3,15).
Función del pediatra de Atención Primaria
Es importante el conocimiento de esta patología por parte del pediatra de Atención Primaria, posibilitando el rápido diagnóstico y derivación al hospital cuando esta sea necesaria.